第三十章 药物动力学
一、概述
(一)药物动力学概念及其临床意义药物动力学(pharmacokinetics)亦称药动学,系应用动力学(kinetics)原理与数学模式,定量地描述与概括药物通过各种途径(如静脉注射,静脉滴注,口服给药等)进入体内的吸收(Absorption)分布(Distribution),代谢(Metabolism)和排泄(Elimination),即A.D.M.E.过程的“量时”划化或“血药浓度经时”变化的动态规律的一门科学。药物动力学是一门较年青的新兴药学与数学间的边缘科学;是近20年来才获得的迅速发展的药学新领域。目前国内对Pharmacokinetics一词的翻译方法颇乱,除称为“药物动力学”、“药动学”之外,尚有称作“药物代谢动力学”、“药代动力学”等名称。总之,该名称的泽法暂未统一,这些译语往往是来自同一概念,指的是同一门学科,请读者不要误解。特别是采用“药物代谢动力学”以及“药物动力学”的作者,他们在该词中指的“代谢(Metabolism)概念是广义性的,包括了药物在体内的A.D.M.E,的整个过程。但考虑到国外在Pharmacokinetics领域中,Mitabolism一词多半都是狭意的概念,仍然仅指生物转化而言,况且在Pharmacokinetics领域中确实存在着“Pharmacokineticsofmetabolism(代谢,即生物转化的动力学)”这一部分内容,而这部分内容显然不能代表整个Pharmacokinetics。著名的药物动力学创建人之一J.G.Wanger有一个很好的说明,他指出,Pharmacokinetics一词,是指将动力学(kinetics)的原理用于pharmakon,而pharmakon一词源出于希腊文意指药物和毒物。在日本国内,一概把此词译作“药动力学”。鉴于我们采用了“药物动力学”作为pharmacookinetics的中译名。药物动力学近年来的发展和应用,日益证明了它在药学领域中所占的特殊重要地位。首先,药物动力学作为一门用数学分析手段来处理药物在体内的动态过程的科学,具有重大的理论价值,是“数学药学”的重要组成部分,它的基本分析方法已经渗放到生物药剂学,临床药剂学,药物治疗学,临床药理学,分子药理学,生物化学,分析化学,药剂学,药理学及毒理学等多种科学领域中,已成为这些学科的最主要和最密切的基础,推动着这些学科的蓬勃发展。同时,药物动力学还有着析为广泛的实用意义,它的发展将对现有的药物的客观评价、新药的能动设计、改进药物剂型、提供高效、速效、长效、低毒副作用的药剂,特别是对于临床指导合理用药,通过药物动力学特征的研究,要挟临床治疗所需有效血药浓度选择最适剂量,给药周期,负荷剂量的计算,以及连续用药是否会在体内发生蓄积,设计最优给药方案等具有重大的实用价值。总之,药物动力学已成为一种新的有用的工具,已被广泛地应用于药学领域中和各个学科,成为医药研究人员和广大医药工作者都需要了解和掌握的学科。联合国世界卫生组织的一份技术报告中曾强调指出:“对评价药物疗效与毒性来说,药物动力学的研究,不仅在临床前药理研究阶段,而且在新药研究的所有阶段都很重要。”尽管对我们来说,不仅在临床前药理研究阶段,而且在新药研究无论是现在还是将来,都有着重要的意义。
(二)药物动力学的发展历史药物动力学的发展仅几十年的历史,国际上于1972年,由国际卫生科学研究中心(InternationalCenterforAdvancedStudyinHealthSciences)的J.E.Fogar发起在美国马里兰洲波兹大国立卫生科学研究所(N.I.H)召开了药理学与药物动力学国际会议,在这次具有历史性意义的会议上,第一次由N.I.H这样的权威性机构正式确认药物动力学为一门独立学科。
早在1913年,Michaelis和Menten就提出了有关动力学方程;1919年,瑞士的Widmark利用数学公式对药物有动态规律进行了科学分析;1924年WidmarkandTandbery提出了开放式单室模型动力学;1937年。Teorell又提出了双室模型动力学的假设,并用数学公式详细描述了双室模型动力学规律,在“国际药效学志”(InternationalArchivesofpharmacodynamics)上发表的题为“体内投用物质的分布动力学”的两篇文章,由于数学公式十分繁杂。这一开创性的工作在当时未得到重视和公认;到了60年代,由于电子计算机的重大发展和分析化学和重大突破(它已使人们能从极少量的生物样液中定量测出痕量的药物和化学物质的浓度)以及许多科学家的远见卓识,使药物动力学有很大发展;70年代初,药物动力学才被国际上公认为独立学科。德、美、日等国的药学家F.H.Dost,E.Kruger-Jhi-emer,J.G.Wagner,G.Levy,E.Nelson,M.Gibaldi,褂见喜一郎,花野学等著名科学家都为创建本学科作出了很大贡献,他们在药物动力学的发展史上占有特殊地位。
70年代以来,药物动力学的研究,在理论上,实验方法上和实践应用上都有了飞速发展。目前,还有人用概率论的随机过程论来研究药物的体内动态过程,“矩”汉已经成功地用来分析药物体内各主要过程的“平均驻留时间”,但这种方法严格讲,已经不依赖于室模型。
近年来,人们已致力于发展一类生理学上逼真的药物动力学模型。这种细致的模型基本上是利用了人或其他动物的已知解剖学与生理学情报以及掺入的生理,解剖及生化测定数据。原则上讲,这种细致的模型在某些方面优于经典的隔室模型。从观念上讲,生理学模型精确的表征了任何器官或组织中药物浓度的经进过程,所以能更明晰地洞察药物在体内的分布状况;同时,由于生理学模型的诸参数均相当于诸如器官血流量及容积等真实的生理解剖学数值,故机功能的生理或病理改变所引起的药物配置动力学的变化,有可能通过某些有关参数的变动来预估;最后,这种模型也为采用“动物类比法”提供了可能性,该法为各类动物之间药物资料的相关关系提供了合理基础。生理学模形的提出、确证和应用,代表着一个非常卓越的研究领域。Bischoff及Pedrick在这一领域的先导性研究及卓越贡献是值得称颂的。从生理学模型和各种细节上说,这种药物动力学方法可能用来洞察复杂的生理学研究。总之,药物动力学在过去数年来,国际上开展了大量研究工作,既有精心设计的实验,也有理论上的探讨,取得了很大的成果,特别是70年代中后期,国际上我采用电子计算机编程序处理,处理的准确性,精度与速度都大为提高。药物动力学的原理与方法,如今已经渗透到药学领域的多种学科之中,越来越显示出它的重要性。但是,目前药物动力学的研究,距离完全把握药物在体内的每个器官的动向,从而任意能动地设计出在体内任何指定部位、指定时间、发挥指定作用的药物与制剂,还有相当大的距离。在我们面前,很多方面还是有待于不断认识的必然王国,我国医药工作者,近年来尽管亦进行了大量的研究和探讨,但是,今后亦应在这一领域中花更大气务,吸收国外比较先进的东西,作多方面的实际研究工作和理论探讨,以利于医药卫生事业的发展。我国科学技术已迎来百花争艳的春天,每秒数百万次的大型计算机已研制成功,大面积集成电路已鉴定投产,可以予期电子计算机技术的飞速发展与普及,必将有力地推动我国“药物动力学”的研究与应用得到蓬勃发展。
(三)药物动力学研究的内容及意义药物动力学研究的意义在于它在药学领域里具有广泛的应用,近年来,药物动力学的研究在理论上,实验方法上和应用上都有了飞速的发展,特别是电子计算机的应用,推动了药物动力学的发展和应用。
1.药物动力学在新药研制过程中的指导意义:回顾药物研究的过程,剖析某些类型药物的化学结构与药物体内过程之间的关系,不级看出药物动力学对于指导药物设计的重要意义。在研制一种新药时,常常希望通过结构改造来达到所期待的要求,但是化学结构改变以后,使药物体内过程亦发生变化。因此,即要弄清疗效关系,亦必须掌握药物的理化性质与药物内过程之间的关系,药物动力学的原理与方法方法广泛的用于新药的研制过程。在设计新的化合物时,参考药物动力学参数,分析药物结构对参数的影响,发现什么基因会改变药物的吸收和处置的动力学过程,从中找出规律,再用以指导新化合物设计,就能发挥药物动力学对新药设计的指导作用。药物结构的改变可以大大改变药物的动力学性质,巴比妥类药物就是一个典型的例子,较小结构变化,即可显著地改变药物的处置过程。因此,合成一系列具有不同作用特点和不同动力学参数化合物,就或找到几个临床所需要的药物。
对于药物的动力学研究,不仅可求得这些药物的动力学参数,便于进行定量的比较,而且对于由于化学结构的变化所引起体内过程改变的影响可做出分析和讨论。例如,邻氯苯甲异恶唑青霉素与双氯苯甲异噁唑青霉素的结构区别,只在氯原子的取代数量上。
临床应用过程中发现,口服剂量相等的情况下,双氯苯甲异噁唑青霉素的血药浓度是邻氯苯甲噁唑青霉素的2倍,即高出1倍。开始认为双氯苯甲异噁唑青霉素较邻氯苯甲异噁唑青霉素吸收好,当平行比较口服与静脉注射后的血药浓度,发现静脉注射与口服相近,所以不能从口服吸收差异来解释。后来,又进行了药物动力学研究,发现双氯苯甲异恶唑青霉素的消除速度常比邻氯苯甲异噁唑青霉素小的多。分别是0.98h—1与1.65h—1。消除速度小,及表示药物在体内消除慢,因而药物在不断吸收过程中,双氯苯甲异青霉素噁唑青霉素可得到一个较高的血药浓度(高出1倍)。从而可以看出,双氯苯甲噁唑青霉素口服后所以血药浓度较邻氯苯甲异噁异唑青霉素高,并不是由于吸收好的原因,而是由于在体内消除慢的结果。如果要知道双氯苯甲噁唑青霉素消除慢是由于结构改造以后影响代谢还是影响肾脏排泄,还需要进一步的药物体内过程之间的关系对于研究新药进行结构改造工作来说具有重要的指导意义。
我们知道,药物的化学结构决定着药物的理化性质,不同结构的药物,其脂溶性,水溶性,酸碱性,解离度,受酶催化?匀生化学反应的难易程度都不同。因此,在新药设计中,不论是制备同型物还是前体药物,通过化学结构的改造,改变了化合物的理化性质,都可以使药物的动力学性质发生改变:如改变药物的脂溶性或水溶性,就可以改变药物的吸收速率和吸收量,肝脏的首过效应,以及药物的生物利用度;改变药物的选择性,就可以改变药物在组织中的分布、结合、活化或失活;改变药物在体内的时间过程,就可以改变药物的消除,及排泄或代泄速率。从药物动力学角度来说,新药设计的目的,就是通过药物化学结构改造,设计出使体内过程符合临床需要的药物,以满足临床治疗的要求。2.药物动力学在中草药有效成份研究中的意义:中草药有效成份的药物动力学研究,是对祖国医药学发掘、整理、提高的一个崭新课题,它面广量大,具有重大的理论和实用意义。近年来我国中草药研究工作取得了很大发展,如水飞蓟种子提取的西利宾的药物动力学研究。西利宾(Silybin)系水蓟素(Silymairin为中草药水飞蓟种子提取的总黄酮)中的主要成份。水飞蓟素已应用于临床,对慢性迁延性或慢性活动性肝炎疗效较好,亦未发现副作用或毒性反应。近年来的研究证明,水飞蓟素还有明显降低血清胆固醇及降低肝脏脂质沉积作用。为使临床用药方案合理化,并进一步探讨其降低脂保肝作用机理,有必要对其药动力学特性进行研究。经雄性大兔静脉注射西利宾100mg/kg动物实验表明,符合开放式双室模型特征。
3.药物动力学在药理学研究中的重要地位:我们知道,药理学(Pharmacology)是研究药物和生活机体相互作用的一门科学。它一方面研究药物对机体的作用;另一方面研究机体对药物的影响。因此药理学常被分为药效动力学(Pharmacodynamics)和药物动力学(Pharmacokinetics)两大部分。早在40年代后期,Brid就发现,药物的药理作用可以用血药浓度来说明。药物的药理作用强度多与作用部位的药物浓度有关。药物在血液中的浓度又常反映作用部位的浓度。药物治疗的关键就在于使用部位药物浓度维持在最低有效浓度以上和最低中毒浓度以下。
然而,体内药物浓度由各种途径的消除,不会总是保持不变的,而是随时间变化而变化,最终从体内消除。为反映这种变化,把血药浓度变化绘成“血药浓度-时间”曲线。有了这一曲线,通过数学模型的处理,可得到各种动力学参数。从曲线中可以确定药物浓度的最大值,达到最大值所需的时间,出现有效浓度和维持有效浓度的时间,计算药物的生物半衰期,反映药物在体内的吸收、分布、代泄和排泄特点等。
根据大量实验研究,得出血药浓度与药物作用关系的下述三个观点:①药物作用与血药浓度的关系比剂量关系更密切;②不同个体要达到相同血药浓度所需剂量有很大差③差正常动物对某种药物和受体部位之间的个体差异很小,血药浓度相同,在不同动物出现的作用亦相似。例如,速尿和利尿作用强度与血药浓度之间存在着密切关系。静脉注射速尿以后,不论是尿流量还是Na+排出量的对数值,都与血药浓度的对数值存在着良好的线性关系。因此,研究不同时间血药浓度的变化,在估价药的治疗作用和毒副作用方面具有重要的意义。又如水杨酸的血药浓度在50~100mg/h出现镇痛作用;大于250mg/h则有抗风湿作用;在350~400mg/h具有抗炎作用;达到500mg/h则出现毒性;当达到1600~1800mg/h则引起中毒死亡。对于一些药物临床上已经肯定了血药浓度与疗效的关系,不达到有效浓度则不出现疗效,而超过一定限度则出现毒性。Winek,Koch-Weser和Vesell等查阅了大最文献资料,收集了许多药物有的效浓度。
4.药物动力学对临床用药的指导意义药物动力学与临床药学相结合,产生了临床药物动力学(clinicalpharmacokinctics),主要是研究实现临床给药方案个体化,包括给药剂量,给药间隔时间,给药途径以及剂型的选择等方面的内容。国外临床药学研究生及临床师的培养计划中必不可少的课程之一。
在临床给药方案设计中,药物剂量的确定,若给药剂量太小,则无效;剂量太大,则容易引起中毒。究竟多大剂量适宜,需要药物动力研究,方能作出正确的回答。又如多剂量给药时,给药间隔时间的确定,给药间隔时间长则不能保持体现内有效的血药浓度;若给药时间过短,不仅用药过繁很不方便(特别是注射给药),还容易造成体内药物蓄积中毒。如果根据药物动力学研究,知道药物的生物半衰期或平均稳态血药浓度或最低稳态血药浓度等参数,则有助于临床医师,药师用动力学方法设计出给药间隔,负荷剂量,维持剂量等科学的给药方案,特别是器官病变患者给药方案设计,通过血药浓度监测实现给药方案个体化,象“量体裁衣”一样,具有重要意义。
药物动力学的理论和参数对临床合理用药的指导作用是多方面的,例如,曾被临床广泛应用的三磺片(ST、SD、SM2各0.167g制成0.5g的片剂,和三磺合剂ST、SD、SM2各取3.3%,制成10%的混悬液)。近年来,经药物动力学研究发现,这三种磺胺联合应用由于三种药物的生物半衰期和血浆蛋白结合率相差悬殊,很难保持体内有效血药浓度。见表30-1。药物的t1/2短,从体内消除的快,t1/2长,所以,很难保持体内有效血药浓度。再则三种磺胺血浆蛋白结合率相差很大,联合应用以后,将发生竞争性结合,结合率大的SM2夺走了血浆蛋白,结合率小的SD,在血液中呈游离状态,达到作用部位后,使其作用强度和副作用大大增强,从以上分析可以看出,三种磺胺联合应用,很难保持有效血药浓度,因此,对此,结其生产和应用价值应重新估价,现已停止使用。
表30-1三种磺胺药的t1/2与血浆蛋白结合率
| 药物名称 | t1/2(h) | 血浆蛋白结合率(%) |
| ST SD SM2 | 4 17 7 | 55~80 45 80 |
5.药物动力不在药剂学、生物药剂学等学科领域中的重要地位:药物动力学与药剂学相结合,产生了生物药剂学(Biopharmaceutics)是研究药物及其剂型在体内的吸收、分布、代谢与排泄过程,阐明药物的剂型因素和生物因素与药效关系的一门科学。其研究目的在于通过制剂的生物药学研究所提供的资料,可以正确地评价药物制剂的质量,设计合理的剂型,制剂工艺为临床合理用药提供科学依据,保证临床用药的安全性的有效性。在长期的临床用药工作中,人们常常发现,同一药物制剂的不同药厂出品,或同一药厂同一制剂的不同批号之间,疗效相差很大。国外也有类似的报道,1968年澳大利亚生产的苯妥英钠片剂,病人服用疗效一致很好。后来,有人将处方中的辅料CaSO4改为乳糖,其它未变,结果临床应用时连续发生中毒事件,是什么原因呢?引起人们特别注意。经药物动力学研究发现,将处方中的CaSO4改为乳糖以后,压制的片剂体外释放和体内吸收都大大提高,使血药浓度超过了最低中毒浓度,因此发生中毒事件。1964年还有篇报道,治疗风湿性关节炎的沷尼松片剂,剂量达到原来的4位亦不显效。经研究发现,无效片剂释放一半所需的时间,即T50为173分钟;有效片剂释放一半,即T50为4.3分钟。但此两种片剂崩解时限为2.5分钟。大量事实证明,片剂崩解了,但药物不一定能够完全释放。片剂释放问题,必然影响药物的吸收和临床疗效。在过去的药典中规定,片剂的崩解时限是只要在规定的时间内,能通过10目筛,即崩解到颗粒小于1.6mm即为合格。但是,大多数药物要以分子状态才能吸收,那么,从1.6mm再继续分散到可以吸收的分子状态,还要经过漫长的过程,药典规定的崩解实验已经无能为力了。因此,近年来,世界各国及我国新药典对片剂和胶囊剂的部分产品都提出了释放度的要求,国外部分片剂还提出了生物利用度的要求。实践证明,“唯有结构决定疗效”的概念,现在看来,已经不完全正确了。因此,如何评价药物的疗效和制剂质量等重要工作,仅仅依靠原有的经验,显然是不够的,必然联系药物动力学的原理与方法,作进一步的研究工作。
70年代中期,药物动力学应用于药剂学以来,首先是在生物利用度和长效制剂的设计方面。有关生物利用度的专著和综述已有不少,美国药学会杂志(J、Am.PharmAssoc)在1975~1976年间,曾连载10多种药品的生物利用度专论,其中包括地高辛、呋喃坦啶、土霉素、四环素、苯妥英钠、沷尼松、氨苄青霉素、氢氯噻嗪、氢沷尼松、药霉素、华法令、保泰松、磺胺异噁唑等。国内近年来也进行了大量工作,如强的松龙片剂与滴丸剂、扑热息痛片剂及四种型比较,阿期匹林片剂及栓剂、氨茶碱片剂及栓啶栓剂、苯妥英钠片剂。核黄素片剂、氢氯噻嗪片剂、SMZ片剂等。生物利用度研究中,按给药次数,有单次给药和多次给药多种;按被测体液分,常用血药浓度与尿药浓度法两种。者需要依据药物动力学的原理对实验作合理设计,并对结果进行统计学处理。
应用药物动力学原理设计长效制剂的综述亦不少。药剂学中亦有收载。根据释药速度的不同、Weuing等将长效制剂分为四种类型:1型:只有一级速度的缓释部分,而无速释成分的制剂;2型:只有零级速度缓释部分,而无速释成分的制剂;3型:含有零级速度缓释部分,同时含有速释成分的制剂;4:含有一级缓释部分,同时含有速度成分的制剂。以上各类长效制剂者有各自的血药浓度与时间的关系式,均为应用药物动力学的方法设计的典型例子。
Chandrasekaran等最近应用药物动力学原理设计控制系统的基本原理及典型例子的综述,详细地叙述了口服控制传递系统、东莨菪碱透皮治疗系统及介绍了微型渗透泵了基本设计参数和工艺。前体药物(Pro-drug)有时可使药物长效化,的来利用药物动力学原理对此加以探讨的例子亦有报导。
综上所述,药物动力学已成为一种新的有用的工具,它在药学领域里具有广泛的应用。医学上一些重大课题,如癌症、冠心病、高血压等迄今尚未找到的疗效卓越的新药。因而,寻找新药的方式,正在逐渐从经验转向更为合理的形式。例如,通过生物化学、生物物理学、酶学、药物动力学、统计学以及各种光谱技术以发展或设计新药、新制剂、新剂型。近年来,很重视化学结构与生物活性间的定量关系的推导,从而设计更为优越的药物。这类方法中,Hansch方程式的应用正日渐增多,但还有许多问题尚未解决,如代谢产物产生的毒性,药物与血浆或组织内蛋白相结合而失去效用,以及药物的立体因素等问题。量子化学的应用尚在初始阶段,尚未能满意地解决结构与活性间的关系。应用数、理化最新技术和药物动力学方法,将为新药研究开辟新的途径。从而创制新药、好药、征服各种顽症、绝症,开创我国医药卫生事业的新局面。
二、临床给药方案和设计
(一)概述80年代医药事来飞速发展,临床应用的药物品种越来越多,据统计,目前国际上现有原料药品约4500余种,并且每年平均增加20~30种。
药物品种日益增加,临床滥用或不合理用药亦日益增多,医药开支日益增大,但临床药物治疗水平在某些方面并没有随着药品品种的增加而有较大提高,由于滥用或不合理用药,临床不断出现严重的医疗事故或引起药源性疾病。
据报导,美国市售药物制剂五十余种左右,经调查发现约有90%属滥用。例如,普通伤风感冒,一开始就使用抗生素就是滥用,因为一则无效;且易产生抗药性;还易引起体内菌群平衡失调,美国FDA成立专门机构,严格控制滥用抗生素。
50年代,德国应用机锡胶囊剂抗感染,结果造成217人中毒102人死亡。
1956年,在西欧市售新药反应停(Thalidomide)治疗妊娠反应,造成8000多畸形胎儿诞生,引起震惊世界的悲惨后果。
1968年有篇报道,澳大利亚生产的苯妥英钠片剂,病人服用疗效一直很好。后来有人将辅料CaSO4改为乳糖,其它未变,临床应用相同剂量,结果连续发生严重中毒事件。后来经生物利用度研究发现,这两种片剂虽然剂量相同,但由于辅料改变引起生物利用度较大变化,使血药浓度发生较大变化导致医疗事故。
还有些药物,治疗剂量与中毒量之间相差很小,每个人对其耐受性和体内消除速率又有很大差异,临床用药稍有不慎则容易产生中毒,甚至死亡。
临床药学是为病人治疗和合理用药之间架设的桥梁,是药理学与药剂学的临床应用,包括病人在用药治疗过程中的临护,使药物发挥有利的一面,尽量减少不利的一面。随着医药卫生事业的飞速发展,药师必须面向临床,必须对制定给药方案的有关知识有较深入的了解,才能在协助医师合理用药方面做出较大贡献。药学教育的根本任务是培养药师,从脱离患者的药学、转向为患者服务的药学,这是无需讨论的当务之急。
1.临床药物动力学与药师的基本任务药物动力学(Pharmacokinetics)与临床药学(ClinicalPharmacy)相结合,产生了临床药物动力学(ClinicalPharmacokinetics),是对每一个患者都能提供安全、有效的治疗方案,包括给药途径、用药剂型、用法、用量、给药间隔等,实行给药方案个体化;可以重新审查给药计划;对不良反应做出定量的解释;对正在进行的血液,腹膜透析患者出现不良反应有助于按计划暂时中止给药及做必要的紧急解毒措施等。
作为临床药师的最基本任务是实现给药方案个体化,进行血药浓度监测的实验设计;数据的统计学处理;受试药剂的制备;广泛收集药学情报;应用临床药学动力学等方面的知识为临床医师提供科学给药方案,做到给药剂量个体化,进一步提高药物的疗效,减少药物的不良反应。
2.给药方案个体化与血浓度监测:
(1)给药方案个体化:目前药理学和治疗学教科书中推荐的药物剂量,大都是平均剂量,就如服装店里所卖成衣的尺码一样。但是,成衣尺码还有不同长短和肥瘦可供顾客选用,而教科书中的剂量,却都是固暄的一个。事实上,只有少数安全、低毒的药物按照既定的平均剂量给药,能使用药者获得满意的疗效。但多数药物并非如此。给予同一剂量后,往往只有一部分病人疗效满意,另外一些病人,或因剂量不足疗效不佳,或因药量过大出现不良反应。有时由于病人体内器官病变,影响到药物在体内的正常吸收、分布、代谢和排泄等动力学变化,即使应用常规剂量,有时或无效或产生中毒,血药浓度监测是帮助实现给药方案个体化的重要手段之一,给药方案个体化则是提高临床疗效的一个重要保证。
(2)血药浓度监测:从医生处方到药物发挥治疗作用或产生不良反应要经过药剂过程(PharmaceuticalProcess)、药物动力学过程(PharmacdynamicProcess)和治疗作用过程(TherapeuticProcess)和治疗作用过程(TherapeuticProcess)等四个过程,因此对药物治疗进行的监测应该包括对上述四种过程的全面监测。
通过治疗药物血药浓度监测,对患者的疾病,所用药物的性质,个体对药物的反应等方面充分了解,借助于特定时间的血药浓度,利用临床药学动力学的原则和公式为病人设计体体化的给药方案,联系临床实际,不断提高临床用药水平。
3.血药浓度监测在给药方案个体化中的地位药理作用强度与剂量在一定范围内密切相关,这是药物学的一条基本规律,是针对群体的平均情况而言。当讨论临床具体病人的处方剂量与所得药效强度之间的关系时,则需考虑下面六个问题:①医生虽然开了处方,但病人是否按医嘱中的给药方案用药?②是否由于使用不同厂家和不同批号的主品、因制剂生物利用度不同而影响疗效?③虽然按处方医嘱用药,生物利用度亦保持一致,但有无可能由于个体病人的药物动力学特点存在个体差异,造成血药浓度的个体差异,从而影响疗效?④虽已按医师的愿望调整并建立了一定的血药浓度,但后者能否反映作用部位的药物浓度?⑤即使控制了作用部位的药物浓度是否一定能保证满意的疗效?⑥显然还要考虑由于其他药物存在而出现协同或拮抗作用的可能性?
以上六个环节都可能使处方剂量与药效强度之间丧失相关性。吸有对这六个环节都有了透彻的了解,才能明确血药浓度监测在给药方案个体化中的地位。
4.哪些情况需要血药浓度监测?在药物浓度一效应关系已经确立的前提下,下列情况需要血药浓度监测:①安全范围较窄的药物,其有效浓度和中毒浓度比较接近,如地高辛、锂盐、茶碱等。②米氏动力学过程的药物,在治疗剂量范围内已呈现零级过种,机体对药物的消除功能已达饱和状态,随剂量增吕,血药浓度不成比例地猛增,伴以消除t1/2明显延长,如阿斯匹林、水杨酸盐、苯妥英钠、普萘洛尔等。③为了确定新药的群体给药方案,进行临床药物动力学研究。④药物动力学的个体差异很大,特别是由于遗传性造成药物代谢速率明显差异的情况,如普鲁卡因胺的乙酰代代谢。⑤中毒症状容易和疾病本身美混淆的药物,如用地高辛控制心律失常时,药物过量也可以引起心律失常。⑥常规剂量也可以引起心律失常。常规剂量下没有看到疗效,测定血药浓度有助于分析疗效不佳的原因(制剂质量有问题,药物消除太快……)。⑦常规剂量下出现毒性反应。⑧药物的消除器官功能受损(如肾功能较差的病人应用氨基糖甙类抗生素;肝功能损害病人应用利多卡因功茶三等)。⑨怀疑由于合并用药而出现的异常反应。⑩诊断的处理过理中毒。
(二)肾功能不良病人给药方案的设计当肾病患者应用某种全部或部分经肾消除,且治疗指数较低的药物(发氨基糖甙类抗生素)时,通常很难掌握既维持疗效,又不产生毒性的给药方案。因为此类药物的t1/2,在肾功能不良患者与正常人比较,t1/2要长的多,按肾功能正常人的给药方案很容易产生中毒和医疗事故。为此,对肾功能不良患者的给药方案需要进行调整。
肾病患者给药方案设计:在临床约药方案设计时,亦希望肾功能不良患者体内也能获得在健康人体内显效的平均稳态血药浓度(Css)水平。
对于肾病患者亦有相似的公式:

式中:Css(r)为肾病患者体内平均稳态血药浓度:F(r)为生物利用度;Xo(r)为给药剂量;Ke(r)为消除速度常数;Vd(r)为表观分布容积;
为给药周期。令
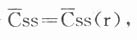
若FVDA与肾功能损害程度无关,则
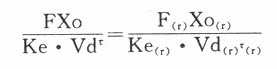
根据此式则肾病患者给药方案调整为:
1.若给药周期不变(τ=τ(r)),肾病患者给药剂量Xo(r)为:

则肾病患者的给药剂量为:
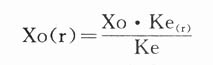
2.若给药剂量不变(Xo=Xo(r)),肾病给药周期τ(r)为:
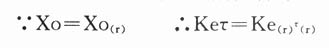
则肾病患者的给药周期为:
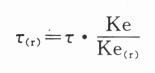
肾病患者的给药方案调整,根据1,2式,只在知道该药物Ke和该患者个体ke(r),即可调整给药剂量给药周期。
对于肾功能减退而ke(r)减小的药物,患者个体消除速度常数ke(r)可以应用Ritschel一点求出或用下面的经验公式求出:ke(r)=a+bclcr式中:clcr为患者的肌酐清除率;a为该药物肾外消除速度常数;b为比例常数。
例如:庆大霉素常用剂量为80mg,给药周期为8h,若测得患者Clcr为40ml/min/m2时,如何调整该患者的给药剂量?
解:查表得:a=2.0b=0.28Ke=30h—1
根据Ke(r)=a+bClcr,则Ke(r)=2+0.28×40=13.2h—1
根据
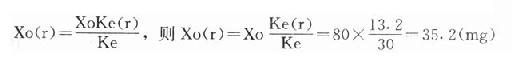
答:该患者给药剂量应调整为每8h给药35.2mg即可。
(三)t1/2与给药方案设计生物半衰期的变动对于每一种具体药物来说,其t1/2并不是一个绝对数值,文献报导t1/2数值正常情况下的平均值,个体有时会有较大差异,多种因素可引起t1/2变动。
1.剂量效应:药物在体内的主动转运,代谢和分布等过程均与药量有关,剂量增加,可能使其中一个或几个过程达到饱和,t1/2将延长,如非线性的药物t1/2随剂量增加而延长。
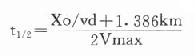
2.尿液的pH的影响从尿中排泄的药物,以肾小球滤过,离子型药物从肾小管排泄,非离子型药物被重吸收,重吸收过程随尿液pH影响,如果药物被重吸收,则一方面延长t1/2,另一方面增加药物在体内的再循环。如伪麻黄碱在尿液pH为5.3时,t1/2为5.0h,pH为8.0时,t1/2为13.0h。
3.个体差异文献报导的药物t1/2为正常人的平均值,个体间存在差Beckett在四个受试者中分别口服右旋苯丙胺15mg的溶液剂,测定体内t1/2分别为4.32h;4.75h;4.93h和5.3h。Kruger-Thiemer报导,磺胺类药物的t1/2个体差异可达2倍左右。
个体差异主要为遗传因素影响,但服用药物进的内环境如饮水量,食物性质及机体活动情况亦有影响。
4.年龄的影响年龄不同,t1/2亦不同,特别是新生儿、早产儿、老年人,临床用药应特别注意。
5.药物相互作用某些药物合并应用时,能增加或降低其它药物的代谢或排泄,使t1/2减小或延长。
例如:双香豆素与甲苯磺丁脲合并服用,可使用甲苯磺丁脲的t1/2从4.9h延长到17.5h,血药浓度增加,使血糖明显下降。
保泰松、丙磺舒、苯海拉明等与其它药物同时用时,可促进自身在体内的代谢使其t1/2缩短。例如强力霉素t1/2这12—16h,若与苯巴比妥合并服用,可使其t1/2大大缩短。
6.疾病的影响与生理因素的影响。
三、老年人药物动力学的特点
老年人对药物的反应与青年人不同,随着年龄的增大,引起人体生理发生变化,从而导致药物动力学的改变。
(一)机体成分的某些变化老年人随着年龄的增加,机体总水分减少,特别是细胞内液,从25岁到75岁,占体重比率从42%降到33%;肌肉组织相对减少;脂肪组织占体重比率相对增加,从18岁到55岁,男性从18%增至36%,女性从33%增到48%。
(二)心脏功能心输出量,随年龄增加而减少,从19岁到85岁,每年的减少将近1%,主要是每搏输出量减少。
(三)肝脏的变化肝血流量,每年递减弱0.3%~1.5%,60岁的老年人约减少40%~50%,肝脏变小,对药物的代谢功能下降。老年人一般多数有高脂血症,动脉硬化的促进因子,促使肝脏脂肪沉积,损伤肝脏功能。
(四)肾脏的变化用对氨基马尿酸测定肾血流量,在40岁以后,呈直线下降,90岁老年人约为20岁青年人的二分之一;肾有效血流量大约每年减少1%;肾小球管分泌功能,随年龄增加而下降,特别是50岁以下降较快,80岁仅达50%左右。
(五)胃肠道的变化男性在50岁以后,女性在60岁以后,胃肠道蠕动开始降低,便秘增多;肠壁血流量减少;空肠,回肠粘膜,肌层萎缩,空肠绒毛面积指数下降,影响吸收;胃酸分泌随年龄增加而减少,胃内pH上升。
(六)其它方面的变化老年人由于蛋白质摄入不足,氨基酸吸收减少,使血浆蛋白下降。27岁的青年人平均为:4.7g/d;65~103岁的老年人平均为3.8g/d。老年人对抗原刺激产生的抗体比青年人减少,免疫力、抗菌力下降;支气管腺毛细胞减少,支气管系统的反应性下降。
四、药物动力学在抗生素临床用药中的指导作用
近年来,在药物作用的研究中,广泛开展了药物动力学的研究,即利用数学模型和公式,对于药物的吸收、分布、转化与消除等过程进行了定量研究。在抗生素的临床前期药理研究中也越来越多的采用药物动力学原理,为抗生素给药方案的制定和合理应用提供参考数据,使抗生素的应用提高到新水平。因此,药物动力学已成为临床工作者日益关心的课题。为了进一步提高今后抗生素的临床药理和临床应用研究水平,就药物动力与抗生素临床应用的关系,特收集了有关文献,资料的报导,整理、介绍给大家在临床用药过程中,或许有一定指导意义。
(一)同类品种药物及药物动力学特性的比较利用各项药物动力学参数,使我们可以对各种药物特别是性质相似的同类品种的体内过程和作用进行定量比较,对临床选择用药及新抗生素的筛选具有一定参考价值。
在耐金葡萄菌所产生的青霉素的半合成青霉素中,异噁唑青霉素类是临床常用的一组,包括苯唑青霉素、邻氯青霉素、双氯青霉素和氟氯青霉素,其药理及药物动力学特性的比较。
由此可见,双氯青霉素和氟氯青霉素对酸最稳定(前者并对金葡萄所产生的β-内酰胺酶较稳定),口服和注射给药后血药浓度较高,肾清除率较低,t1/2较长,结合其在体外产生β-内酰胺酶的金葡萄较增,因此作为口服制剂,优于其同类品种。而且上述两者中,又以氟氯青霉素的血清蛋白结合率降低,在剂量相同时其游离血药浓度较高,t1/2,在静脉给药时对于血管外的刺激小,因此从药理特性看,氟氯青霉素在本组织中无论口服或注射给药,具有更多的优点。
在临床常用的头孢菌素族中,头孢唑啉除了在体外实验中具有抗菌谱广、抗菌强的特点外,从药物动力学参数比较,具有血药浓度高、血半衰期较长,因此药-时曲线下面积较大,体内消除慢,肾清除率低,胆汁浓度高等优点,虽然其蛋白结合率较高,表现分布容积略小,但仍可认为是同一类品种较优良的一种。
国产硫脒头孢菌素(Cefathiamidine)与常用头孢菌素比较,其药物动力学特性与头孢菌素Ⅱ相近。硫脒头孢菌素的半衰期与头孢菌素Ⅱ相差不多,其吸收速率略低,分布容积略小,而药-进曲线下面面积则较大。经初步临床试用,获得良好疗效,准备扩大临床应用。
合用丙磺舒对于硫脒头孢菌素药物动力学过程的影响:合用丙磺舒1.0g后12h内硫脒头孢菌素的尿排泄量显著减少(由排出给药量的93.8%减少为65.7%),同进血药峰浓度显著增加,药-时曲线下总面积则增加非常显著,合用丙磺舒0.5或1.0克后,t1/2均有延工,但不显著。因此,在临床应用中硫脒头孢菌素与丙磺舒合用,可望提高疗效,对于病情较重或致病菌并非高度敏感者,值得考虑采用。
(二)制订合理的给药方案按作用原理的不同,抗菌药物可分为菌剂和抑菌剂两大类。抑菌剂只是暂时抑制细菌的生长,后者的最后消灭依赖于机体的防疫免疫功能,在条件适宜时处于抑制状态的细菌仍有可能恢复活力而继续繁殖。因此,为确保疗效,在采用抑菌剂时,应使血液和体液内达到并保持一定的最低有效浓度,而细菌的最低抑菌浓度(MIC)可以作为衡量最低有效血药浓度的粗略指标。药物以恒速静脉滴注时,经4~5个t1/2,血药浓度可基本达到平衡状态,此时的血药浓度称为坪浓度,使坪浓度达到或超过最低有效浓度,才能保持疗效。在静脉恒速滴注时,坪浓度的高低与单位时间内滴入的药量也就是维持剂量有关而这一剂量可能通过动力学的原理计算出来。
但临床上大都采用间断给药的方式,而在间断给药进(口服或肌注)浓度便不可避免的会出现波动,如果每天的剂量固定不变,则给药间隔越长,血药浓度环绕平均值的波动越大,其低峰血浓度便有低于最低有效浓度的危险。但给药间隔过短,给药次数过于频繁,又影响病人休息,并给实际工作带来困难。实践证明,当给药间隔不超过药物的半衰期(≤t1/2),则平均状态时血药浓度之比不超过2倍,在多数情况下是临床上可能接受的。但在抗生素的应用中,一般都要求血药浓度尽早达到有效抗菌水平,此时可以加用首次冲击量或负荷剂量,这一剂量也可以通过计算求得。
例如:口服土霉素250mg可达到有效抑菌浓度,其半衰期为8h,则其给药方案可定为首剂500mg,维持剂量8h250mg。
由于药物动力学的计算往往非常繁复,不易被一般临床工作者所掌握。Dettli等根据过去沿用的计算公式加以简化,考虑了药物的最低抑菌浓度,分布容积,清除率及血清蛋白结合率等因素,对几种常用磺胺药,用药物动力学的原理进行计算,求得各药物的首次冲击量,维持剂量和给药间隔。
从表中计算所得的给药方案与临床常规用法相比,可能一尽一致,但具有参考价值。特别是其中关于磺胺嘧啶的给药方案有用量减少,给药间隔延长的趋势,与我们的观察相一致。上述方法在合理用药及新药的临床试用中,有助于迅速确定一个较为合理的给药方案。但由于各种复杂的病理过程,个体之间的差异,以及计算中还有许多复杂因素,上述结果是否与临床情况符合,有待实践验证。
(三)预测毒性反应的发生氨基糖甙类抗生素为临床处理各种革兰氏阴性杆菌感染治疗的重要药物,但该组药物对肾脏和神经系统有较显著的毒性,据报导,用庆大霉素治疗的病人产生重肾毒性反应者约为3%~5%,近年来,Coiburn等的研究发现在庆大霉素疗程中,产生肾毒性症状与用量过大但未产生肾毒性症状者其血药浓度均有异常增高,此种增高现象持续至停药后8h此后则前者的血药浓度仍保持在较高水平,而后则迅速下降。因此,血药浓度的测定必须继续进行至停药后24小时,才能区别肾中毒的患者。但根据疗程中定期血药浓度测定的结果,产生肾毒性症状的患者其分布相曲线斜率(a),转运速度常数(K12)以及分布容积等数值显著增高,药物动力学参数表明给药后药物迅速从中央室进入周边室(主要地肾组织中积聚),同时其消除速度常数则随给药剂量的增多而持续下降。这些变化在肾毒性症状出现以前即以相当明显,因此可用以预测中毒症状的发生,对于临床安全用药有较大的参考意义。
肾功能减退时,许多药物的半衰期常显著延长,因此在采用对于肾脏毒性较大及主要经肾排泄的抗生素和氨基糖甙类及多粘菌素等时,剂量及给药间隔应作相应的调整。临床上一般根据血浆肌酐值的变化来推算,但内生肌酐清除率可以更准确的和定量的反应患者的肾功能状态。
根据公式
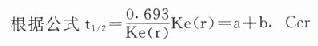
式中:Ccr为内生肌酐清除率,正常人以100ml/min计算;a为每小时非经肾消除速度常数;b为每小时经肾消除速度常数;Ke(r)为每小时总消除速度常数。

例:某病人将采用卡那霉素,其内生肌酐消除率为40ml/min查表:病人的Ke(r)=10.6h--1Ke=25h--1
病人的每日剂量1×10.6/25=0.42
分两次肌注。
或查表:卡那霉素正常人的半衰期为2.75h,
病人的半衰期=69.3/10.6=6.54(h)
患者给药间隔=12×6.54/2.75=28.56(h)
此外,一些主要由肾排泄的抗生素如氨基糖甙类及孢菌素,其总的消除速度常数与内生肌酐清除成直线关系。因此都可以根据不同的肾功能情况来计算和调整给药方案。
一些氨基糖甙类抗生素的总消除速度常数(Ke):
妥布霉素Ke=0.028±0.0025Ccr里杜霉素Ke=0.0147±0.002Ccr
西梭霉素Ke=0.0052±0.0024Ccr丁胺卡那霉素Ke=0.0094±0.0034Ccr
尼蒂霉素Ke=0.017±0.002Ccr
常用头孢菌素类的总消除速度常数:
头孢菌素Ⅱ,Ke=0.067±0.003.Ccr
头孢菌素Ⅳ,Ke=0.0355±0.0062.Ccr
头孢菌素Ⅴ,Ke=0.014±0.003.Ccr
头孢菌素Ⅶ,Ke=0.008±0.006.Ccr
头孢菌素Ⅷ,Ke=0.184±0.007.Ccr
从上式求出在不同肾功能时的Ke值后,代入下式,即可得出患者应采用的剂量或给药间隔。

近年Hull与Sarubbi二氏根据庆大霉素血药浓度测定的结果用电子计算机算出各项药物动力学叁数及在一定剂量时预期可能达到的血药浓度;制成在不同程度肾功能减退时,为达到预期的血高峰浓度所需的庆大霉素剂量。
例如:患者体重70kg,内生肌酐清除率为18ml/min,似采用庆大霉素,该药对于细菌的最低抑菌浓度为1~2ug/ml,需达到的血药浓度为4~6ug/ml。
从“庆大霉素首次冲击量”表查出,为达到4~6ug/ml的血药浓度,应给予首次冲击量。1.5mg/kg×(10-7)=94mg即按去脂肪后体重计算(扣除体重的10%),为达到上述血浓度应给予首次冲击量94mg。再查表中肌酐清除率为18ml/min时,每12hr给予首次冲击量的55%,即94mg×55%=52mg,即可保持上述血药浓度。但肾功能不稳定的患者,其维持剂量应根据每天的内生肌酐清除率进调整。随着治疗的进行,氨基糖甙类药物可在肾组织中逐渐积聚并超过血肌酐值所能反应和程度。因此,在给药3~4天后应进行血药浓度测定,后者增高预示毒性反应易于发生。
(四)生物利用度(Bioavailability)的测定临床实践发现同一种药物由于其剂型、配方及工艺等不同,口服和肌注后可以显著影响吸收,因而影响临床疗效。1968年美国有关当局收回了8种市售不同配方的氯霉素胶囊2亿粒,这些药品全部符合药典和美国食品药物管理局(FDA)规定的质量标准,但以临床应用证实吸收不良,生物利用度差。因此近十年来,药品的生物利用度问题在国内外已日益受到重视。
生物利用度涉及药物的吸收过程与吸收速率两方面,药物的吸收速度与吸收程度不一定有联系,吸收快的未必一定吸收完全。肌肉注射并不一定口服者吸收快或吸收完全。例如磺胺药就是一个例子,肌肉注射磺胺药后血药浓度高峰并不提早到达,其血药浓度也未见于相同剂量口服者。在抗生素的临床应用中,通常采用首次冲击量或经静脉给药,以保证血药浓度迅速达到有效水平。
通常测定某一药浓度经口服或肌注后的生物利用度必须测定其给药后的药一时曲线下面积与静脉给药者相比,也可以用公式计算来比较同一药物两种不同配方的生物利用度,但这些方法的缺点是在实验过程中须多次抽血,Barr氏等比较三种不同配方口服四环素盐酸的生物利用度,他们发现单次给药后48~72h尿药排泄量计算所得的药物动力学参数可以比较两种不同配方的药物生物利用度,其优点是可能免去多次抽血。同时,根据单次给药后血药浓度测定计算所得参数可以预测两种不同配方四环素胶囊的生物利用度的差异更加容易显示出来。例如一次服用250mg时,两种四环素制剂的生物利用度几乎没有显著差别,但一次服用500mg或750mg时,其中差别就清楚的显示出来了。这一事实说明了临床服用四环素时,如不经生物利用度试验,每日4次,每次250mg的疗效与每日两次,每次500mg者并不一不定期相等,因为不同剂量时的吸收程度程度不可能不同,值得注意。我们曾对国产不同晶型无味氯霉素的生物利用度进行试验,结果证实口服A晶型后血药浓度低于最低有效水平。正常人口服B型后的血药浓度较A型者显著为高;但B型非微粒加吐温-80后血药浓度可显著提高,超过B型微粒型而与口服等量氯霉素者相近。在目前工艺条件尚不能生产微粒型时,采用B型非微粒型加入适量吐温-80,可使血药浓度达到有效水平,保证临床疗效,从而收到了较大的经济效果。
国内曾对四环素、土霉素的碱及盐酸盐进行生物利用度比较,但由于各地的试验方法尽不相同,数据分析处理不够精细,因此所得结果参差不齐,未获肯定结论。这一工作对于临床治疗有较大意义,建议今的继续研究。
综上所述,根据药物动力学的原则,可以用数字来描述药物的体内过程。从血药浓度通过计算可参得到更多信息,使我们对药物的药理特性有更详尽的了解,并可以进行定量比较,有助于制订合理的给药方案,根据机体情况调整给药方案,预测毒性的发生等。但也应看到,由于人体的复杂性,无论通过如何细致复杂的计算方法,药物动力学研究只能得出一个大致的估计;在此基础上如能辅以细致的临床观察和其他测试方法,则可使些项研究更好地为临床服务,最大限度地发挥药物治疗效果,并促进合理用药。
-
《医院药学》 中的相关章节:
……
第二十五章 中药房的质量管理
第二十六章 药学科的经济效益管理
第二十七章 药品的淘汰
第二十八章 临床药学概述
第二十九章 临床药理学概述
第三十章 药物动力学(当前页)
第三十一章 生物药剂学
第三十二章 药效学研究
第三十三章 抗生素的合理应用
第三十四章 中药的临床药学
……