第126节 代谢性和先天性肾脏疾病
肾脏疾病可能源于解剖或代谢异常,很多是遗传性并在出生时就存在(先天性)。
肾小管酸中毒
肾小管酸中毒是肾小管不能充分将血中的酸分泌入尿中的一种疾病。
正常情况下,肾脏清除血中的酸并将其泌入尿中。肾小管酸中毒时,肾小管不能正常行使功能,使泌入尿中的酸量减少,其结果酸潴留在血中,发生代谢性酸中毒(见第138节),从而导致下列异常:
・血钾浓度降低
・肾脏钙质沉着
・脱水倾向
・痛性骨软化和弯曲(骨质软化或佝偻病)
肾小管酸中毒的病因可能是遗传性、药物、重金属中毒或自身免疫性疾病,如系统性红斑狼疮或干燥综合征(斯耶格伦综合征)。
肾小管酸中毒的类型
| 类型* | 原因 | 基本异常 | 引起的症状和代谢异常 |
| 1 | 遗传性;自身免疫性疾病或某些药物;常常原因不明,尤其是妇女 | 小管不能分泌酸入尿中 | 血酸度高;轻度脱水;低血钾;导致肌无力和瘫痪;易于骨折;骨痛;肾结石(钙质沉积);肾衰竭 |
| 2 | 常是遗传性疾病引起,如范可尼综合征(Fanconi综合证)、遗传性果糖不耐受或脑-眼-肾综合征(Lowe综合征);也可能由重金属中毒或某些药物引起 | 小管吸收碳酸氢盐障碍以致尿中碳酸氢盐丢失 | 血酸度高,轻度脱水和低血钾 |
| 4 | 非遗传性疾病;由糖尿病、自身免疫性疾病、镰形细胞病、尿路梗阻引起 | 对调节肾脏钾、钠排泄的一种激素――醛固酮无反应或反应低下 | 血酸度高;高血钾,除非血钾过高引起心律紊乱和肌瘫痪,很少引起症状 |
*注:无3型
【症状和诊断】
有三型肾小管酸中毒,每型产生略有不同的症状。血钾浓度降低时,可发出神经症状,包括肌无力、反射减弱,甚至瘫痪。也可发生肾结石引起肾脏细胞损伤,导致肾衰竭。
如果病人具有某些特异性症状或血常规检查有高酸和低钾时,医生应考虑到肾小管酸中毒的诊断。一些特殊检查有助于确诊肾小管酸中毒的类型。
【治疗】
治疗取决于类型。1型和2型每日口服碳酸氢钠(食用苏打)溶液以中和血中的酸,这种疗法可以解除症状,预防肾衰竭和骨病或防止上述临床表现恶化。其他特殊制备的溶液也可以使用,补充钾也可能是必要的。4型酸中毒较轻,一般不需碳酸氢钠治疗,足量饮水和口服利尿剂可以防止高血钾。
肾性糖尿
肾性糖尿(葡萄糖尿)是血中葡萄糖浓度正常或低下情况下尿中仍有葡萄糖排出的一种疾病。
肾脏有血液滤过器的作用。血液流经肾脏滤过时,葡萄糖同其他许多物质一道被滤出。滤液通过肾小管网时,需要的物质,包括葡萄糖,被重吸收入血中,而不需要的物质被排入尿中。大多数健康人,葡萄糖完全重吸收入血中。
正常情况下,仅当血中葡萄糖过多时,人体才会排泄葡萄糖入尿中。而肾性糖尿病人,即使血糖水平正常,尿中也有葡萄糖排出,这种糖尿的发生,是由于肾小管功能异常。肾性糖尿可能是一种遗传性疾病。
肾性糖尿无症状,对人体也无严重影响。如果血中葡萄糖水平正常,尿常规检验发现尿中有葡萄糖,医生即可作出诊断。虽然偶尔肾性糖尿病人会发生糖尿病,但是一般不需治疗。
肾源性尿崩症
肾源性尿崩症是因肾脏对抗利尿激素无反应和不能浓缩尿液而产生大量稀释尿的一种疾病。
肾源性尿崩症和人所共知的糖尿病(见第147节),均可排出大量的尿,除此而外,两种疾病是完全不同的。
【病因】
正常情况下,肾脏根据人体的需要调节尿的浓度,肾脏作出这种调节是取决于血中抗利尿激素的水平,抗利尿激素由垂体分泌,其作用是使肾脏保水和浓缩尿。
有两种类型尿崩症存在。肾源性尿崩症是由于肾脏对抗利尿激素无反应,所以肾脏持续排出大量稀释的尿;另一种类型是垂体不能分泌抗利尿激素(见第144节)。
肾源性尿崩症可能是遗传性疾病。引起这种疾病的基因是隐性的,携带在X染色体上,所以通常仅男性发生症状,而携带有这种基因的女性能将这种病遗传给其儿子(见第2节图)。其他肾源性尿崩症的原因包括使用某些可损害肾脏的药物,如氨基糖甙类抗生素、去甲金霉素以及治疗躁狂抑郁症的锂。
【症状和诊断】
遗传性肾源性尿崩症通常在出生后立即出现症状。这些症状是极度口渴(烦渴)和排出大量稀释的尿(多尿)。因为婴儿不能表达口渴,结果造成极度脱水,患儿可以发生高热,伴以呕吐和惊厥。
若肾源性尿崩症得不到及时的诊断和治疗,可以损伤脑,遗留永久性精神迟钝后遗症。频繁的脱水发作也可延迟身体的发育,但是患这种疾病的婴儿,经过治疗,发育可能正常。
根据上述症状,医生作出肾源性尿崩症的可疑诊断,实验室检查显示血钠浓度增高和极度稀释的尿。除此而外,肾功能基本正常。禁水试验检查肾脏对抗利尿激素的反应可证实诊断(见第144节)。
【治疗】
只要肾源性尿崩症病人感觉口渴,就必须常饮适量的水以预防脱水。婴儿和幼儿必须经常喂水。病人饮水足量就不会发生脱水,但是长时期不饮水(一般超过12小时)可导致严重的脱水。某些药物,如噻嗪类利尿剂(如双氢氯噻嗪)和非固醇类抗炎药(如吲哚美辛或托美丁)可能有效。
胱氨酸尿
胱氨酸尿是一种罕见的疾病,由于胱氨酸排入尿中,常常在尿路形成胱氨酸结石。
胱氨酸尿是肾小管的遗传性缺陷所致。引起胱氨酸尿的遗传基因是隐性的,因此这种疾病必须具有两个异常的遗传基因,分别来自父、母双方。携带这种基因但未患本病的个体具有一个正常和一个异常的基因。这些基因携带者尿中可排出比正常人量多的胱氨酸,但很少形成结石。
【症状和诊断】
在膀胱、肾盂(尿收集和流出肾脏的区域)或输尿管(从肾脏输送尿到膀胱的细长管道)形成胱氨酸结石。一般在10~30岁开始出现症状,常常第一个症状是因结石嵌顿入输尿管,输尿管痉挛所引起的剧痛。结石阻塞尿路可引起尿路感染和肾衰竭。
对反复发生肾结石的病人,医生就应检查胱氨酸尿。显微镜下胱氨酸在尿中可形成棕黄色六角形结晶。通过几种试验也可发现尿中过量的胱氨酸。
【治疗】
治疗包括保持尿中低浓度的胱氨酸、防止胱氨酸结石形成。为保持尿中低浓度的胱氨酸,就必须饮足量的水,以维持尿量至少每日4000ml。但是,在夜间由于病人不饮水产生少尿,较易形成结石。就寝前饮水可减少这种危险;另一种治疗方法为服用碳酸氢钠和乙酰唑胺碱化尿,在碱性尿中胱氨酸比在酸性尿中更易溶解。
如果经上述措施仍继续形成结石,可试用青霉胺,青霉胺和胱氨酸反应以保持其溶解。但是约半数服用青霉胺的病人发生副作用,如发热、皮疹和关节疼痛。
范科尼综合征
范科尼综合征(Fanconi综合征)是一种罕见的肾小管功能障碍性疾病,导致尿中过量的葡萄糖、碳酸氢盐、磷酸盐和某些氨基酸排出。
范可尼综合征可能是遗传性的,也可能由重金属类或化学制剂的使用,维生素D缺乏,肾移植,多发性骨髓瘤或淀粉样变性引起,服用过期的四环素也可引起范可尼综合征。
遗传性范科尼综合征症状通常开始于婴儿期,病儿可能有多尿、其他症状有无力和骨痛。
症状和血液检查显示血酸度高,提示医生应怀疑范科尼综合征,如果尿检查发现高水平的葡萄糖、磷酸盐、碳酸氢盐、尿酸、钾和钠,就证实本病诊断。
范科尼综合征是不能治愈的。饮用碳酸氢钠溶液可中和高血酸度(酸中毒);低血钾需口服钾补充剂,骨病需口服磷酸盐和维生素D补充剂治疗。若患儿发生了肾衰竭,应进行肾移植以拯救生命。
维生素D抵抗性佝偻病
维生素D抵抗性佝偻病是一种血磷浓度及血活性维生素D含量低下所引起的痛性骨软化利易于弯曲的疾病。
这种非常罕见的疾病几乎都是遗传性的,由携带在X染色体上的显性基因遗传(见第2节)。基因缺陷引起肾脏异常,促使磷酸盐排入尿中,导致低血磷浓度。因骨的生长需要磷酸盐,它的缺乏导致骨的缺陷。维生素D抵抗性佝偻病的女性病人骨病的严重程度比男性病人轻。罕见的病人,这种疾病的发生是某些肿瘤的后果,如骨巨细胞瘤、肉瘤、前列腺癌和乳腺癌。维生素D抵抗性佝偻病与维生素D缺乏引起的佝偻病是不同的(见第135节)。
【症状和治疗】
维生素D抵抗性佝偻病通常在出生后第一年发病。其症状轻重不等,轻度无明显症状,重度发生弓形腿及其他骨畸形,骨痛和身材矮小。肌肉附着处骨的骨赘可限制相应关节的活动。婴儿颅骨早闭,导致惊厥。实验室检查显示血钙浓度正常,血磷浓度降低。
治疗的目标是提高血磷浓度,促进正常骨质的形成。可以口服磷酸盐,同时应配用活性维生素D--骨化三醇。单独口服维生素D无效。在某些成年病人,癌肿切除后,由癌肿引起的佝偻病显著好转。
哈特纳普病
哈特纳普病(Hartnup病)是一种罕见的遗传性疾病,由于色氨酸和其他氨基酸小肠吸收减少和大量排入尿中,从而引起皮疹和脑异常。
哈特纳普病病人具有两个隐性遗传基因,分别来自父母。这种疾病影响机体制造一些蛋白质的氨基酸加工。病人不能将色氨酸转化为B族维生素的烟酰胺,因而病人不能从小肠吸收适当量的氨基酸并排出过量的氨基酸入尿中,以致体内氨基酸含量减少。
【症状】
阳光、发热、药物、情绪或精神紧张都可诱发症状。发病前,病人几乎都有一段时期营养不良。随年龄增大,疾病发作频繁程度降低。大多数症状散在发生,且由于烟酰胺缺乏引起。皮疹发生于暴露到日光的皮肤,精神迟钝、身材矮小、头痛、步态不稳、虚脱或晕厥常见,可出现烦躁不安。
【诊断和治疗】
尿标本实验室检查发现尿中氨基酸及其降解产物增多。
维持充分的营养,饮食中补充烟酰胺和烟酸可防止哈特纳普病人病情发作;高蛋白饮食可补充胃肠道吸收不良和尿中氨基酸丢失过多引起的氨基酸缺乏。
巴特尔综合征
巴特尔综合征(Bartter综合征)是一种肾脏过多排出电解质(钾、钠和氯),从而导致低血钾浓度(低钾血症)、高醛固酮和高肾素血症的一种疾病。
巴特尔综合征通常是隐性基因遗传性疾病,因此,这种疾病的病人具有这种疾病的两个遗传基因,分别来自父母。
【症状】
巴特尔综合征的患儿生长缓慢,表现营养不良,可有肌无力和极度口渴,多尿,智力发育迟缓。
血中氯化钠浓度和水分降低,机体代偿性产生较多的醛固酮和肾素,这些激素则降低血钾浓度(见第137节)。
【诊断和治疗】
根据症状医生作出巴特尔综合征的可疑诊断,实验室检查结果发现血钾和激素浓度异常则支持本病诊断。
口服钾盐补充剂和减少尿排钾的药物,如螺内酯(也阻断醛固酮的作用)、氨苯蝶啶、阿米洛利、普萘洛尔或吲哚美辛可预防本病的许多并发
症。为代偿过多水分丢失,则应足量饮水。
利德尔综合征
利德尔综合征(Liddle综合征)是一种罕见遗传性疾病,本病肾脏排泄钾,但是潴留大量的钠和水,导致高血压。
肾脏的异常引起利德尔综合征,可以口服氨苯蝶啶或阿米洛利等药物以预防钾排出,增加钠和水的排出,降低血压。
多囊性肾脏疾病
多囊性肾脏疾病是一种双肾多数囊肿形成的遗传性肾脏疾病,双肾长得较大,但仅有少许功能性肾组织。
多囊性肾脏疾病,双肾多数囊肿形成。囊肿逐渐长大,使肾脏部分或大部分肾组织破坏。
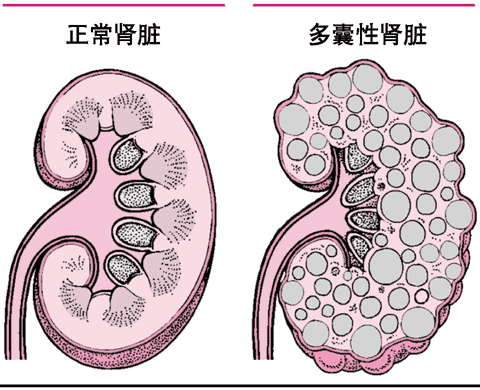
引起多囊性肾脏疾病的基因缺陷可以是显性的或隐性的。换言之,一个本病病人具有一个亲代的显性基因或分别来自双亲的两个隐性基因。显性基因遗传的病人直到成年才出现症状;隐性基因遗传的病人在童年就有严重的疾病。
【症状】
在儿童,多囊性肾脏疾病引起双肾极度长大,腹部凸出。病情严重的新生儿,由于胎儿肾衰竭导致双肺发育不良,可出生后短期内即死亡。肝脏也可以受累,本病患儿在5~10岁有发生连接肠道和肝脏血管(门脉系统)内高血压的倾向,最终发生肝衰竭和肾衰竭。
在成人,多囊性肾脏疾病在多年时间缓慢进展。典型的症状开始于青年或中年期,但也有少数病人直到死亡后尸体解剖时才发现。常有的症状包括背部不适或疼痛、血尿、感染和因肾结石引起剧烈地痉挛性疼痛(绞痛)。另一些病人,由于功能性肾组织减少,可出现乏力、恶心、尿量异常和其他肾衰竭后果。常见的慢性感染可使肾衰竭恶化。约半数多囊性肾脏疾病的病人,在确诊时有高血压。
约1/3多囊性肾脏疾病的病人肝脏也有囊肿,但是这些囊肿并不影响肝脏功能。20%以上的本病患者有颅内血管扩张,其中75%的病人,最终发生脑溢血(蛛网膜下腔出血)(见第74节)。
【诊断、预后和治疗】
根据家族史和症状,医生应怀疑本病。当疾病到晚期且肾脏极度长大时,诊断无疑。超声波扫描和计算机断层扫描可显示囊肿引起的双肾和肝脏特征性的"蛀蚀"样外观。
半数以上的病人在其生命中的某一时期发生肾衰竭。治疗尿路感染和高血压可延长寿命,最终需进行透析或肾移植以拯救生命。
基因咨询有助于多囊性肾脏疾病病人了解其子女遗传本病的可能性。
髓质囊肿疾病
髓质囊肿疾病是一种肾衰竭与双肾深部囊肿同时发生的疾病。
髓质囊肿疾病是一种遗传性或天生缺陷性(先天性)疾病。
症状通常发生于20岁前,但是症状差异非常大,一些病人很晚才出现症状。正常情况下,抗利尿激素使肾脏浓缩尿,本病病人肾脏对抗利尿激素无反应,产生大量的尿,这就可引起大量的钠排出,因此病人必须每日摄入大量的液体和盐(钠)。儿童生长发育迟缓,骨病发生率高。对多数病人而言,上述临床表现在数年内缓慢发生,由于机体代偿良好,直至肾衰竭晚期,才认识上述临床表现。
实验室检查显示肾功能不良。X线检查双肾缩小,超声波扫描可发现双肾深部多发性囊肿,囊肿太小时,超声波扫描可能发现不了囊肿。
本病进展虽缓慢,但却是进行性的。肾衰竭发生后,应进行透析和肾移植。
髓质性海绵肾
髓质性海绵肾是一种充盈尿的肾小管扩张,导致双肾组织呈海绵状的先天性疾病。
髓质性海绵肾病人可持续很长时间而无症状,但易于发生肾结石性肾绞痛、血尿和肾脏感染,半数以上病人发生肾脏钙质沉着。
出现上述症状病人,医生应考虑进行X线检查,X线检查可发现肾脏钙质沉着。静脉注射X线照片可显影的不透射线的造影剂,当造影剂通过肾脏排泄时,在X线下进行观察,这种影像学技术可证实诊断。超声波扫描可协助诊断,但不能发现位于肾脏深部的微小囊肿。
髓质性海绵肾若无肾脏钙质沉着,一般不需治疗。口服噻嗪类利尿剂,大量饮水,进食低钙饮食可以预防结石形成和尿路结石梗阻。若有尿路梗阻,则需手术治疗。感染时,则用抗生素治疗。
遗传性肾炎
遗传性肾炎(Alport综合征)是一种遗传性疾病,临床表现为肾功能减退、血尿,有时发生耳聋和眼病变。
遗传性肾炎的致病基因在X染色体上(见第2节),但是其他因素可影响具有本基因病人病情的严重程度。女性病人的2条X染色体仅一条有致病基因,尽管病人的肾功能比正常人差,但一般无症状;具有致病基因的男性(男人无另一条X染色体以代偿本基因缺陷)通常在20~30岁发生肾衰竭。很多病人除血尿外,无其他症状,但尿中也可含有不同数量的蛋白质。显微镜下可见白细胞、各种类型的管型。
除肾脏外,本病尚可影响其他器官。常见听力下降,通常为高频音阈性耳聋;白内障也可发生,但比听力下降少见。角膜、晶体或视网膜异常有时可致盲。其他临床表现包括多发性神经病和血小板减少(见第155节)。
发生肾衰竭的病人需进行透析或肾移植。对希望生育的遗传性肾炎病人应进行遗传咨询。
甲髌综合征
甲髌综合征(Nail-Patella综合征)是一种罕见的遗传性结缔组织疾病,本病导致肾脏、骨骼、关节和指甲异常。
大多数本综合征的病人有一侧或双侧膝盖骨(髌骨)缺失,一侧前臂骨(桡骨)肘关节脱位,盆骨畸形。病人无指甲或指甲发育不全,有凹陷和凸起。眼虹膜可出现异色。
可能有蛋白尿,通常小量,血尿罕见。若有血尿,医生应进行肾功能检查。约30%肾脏受累的病人最终发生肾衰竭。骨X线检查和骨活检可证实诊断。
大多数病人不需治疗。若病人发生肾衰竭,应透析和肾移植。希望生育的病人应进行遗传咨询。引起甲髌综合征的基因是显性的,即本病病人的下一代,本病基因有50%的遗传机率。
-
《默克家庭诊疗手册》 中的相关章节:
……
第十四章 肾脏和尿路疾病
第122节 肾脏和尿路生物学
第123节 肾衰竭
第124节 肾炎
第125节 肾脏血管疾病
第126节 代谢性和先天性肾脏疾病(当前页)
第127节 尿路感染
第128节 尿路梗阻
第129节 神经源性膀胱
第130节 尿失禁
……